
Introduction – Genomic Variants
Genomic variants are differences from one genome to the next. These variants can take multiple forms, from modifications to a single nucleotide, or the deletion, insertion, copying, translocation, inversion, etc., of entire segments. While a genomic variant can technically be relative to anything, in human genetics we generally talk about genomic variants in one of two contexts:
- comparison between individuals or groups of individuals to a Genomic Assembly, for example, GRch38.
- comparison between the genome of tumour tissue to normal tissue.
These genomic variants are important to study because combinations of them have been implicated in poorer patient prognosis. Genomic risk factors, such as the ones found in 23andme reports, are found by assaying for known variants (usually Single Nucleotide Variants (SNPs)).
Part of what CanDIG wants to enable is making sure genomic data collected by the Marathon of Hope Cancer Centres Network (MoHCCN) is available to researchers.
Data Discovery
CanDIG is a data discovery platform, which means that rather than being interested in just being a store of information, our goal is to enable institutions and consortia to safely share information with authorized researchers. Moreover, CanDIG is deployed at sites, rather than being a centralized store, which allows each local site team to make accessible whatever data they choose for querying and analysis within the federation of CanDIG servers. Due to this federated approach to deployment:
- Researchers can still find if there are patients that fit their criteria by asking the federation
- Hospitals have complete control over their own databases
- Patients can ensure that their data is only used according to the data release policies they have signed off on.
GA4GH’s Beacon protocol propagates queries (such as whether or not there is a specific nucleotide at a specific region on someone’s genome) across a variety of sites that implement Beacon and receive back everybody that matches. CanDIG implements parts of this Beacon protocol in a performant manner.
Data Ingestion
Currently, HTSGet ingests data by creating Data Repository Service objects for each ingested file, which contains metadata about the data for future lookup. We start by considering the entire genome as 10kbp bins, for easier lookup. With the genome being 3.2x10^9 bp in the hg38 assembly, we only have to consider 3.2x10^5 bins, which is a good size for our index. Both these indexes and a mapping for each ingested file’s header to its associated file are stored in a SQLite database.
Suppose we have a region of the genome and two VCFs containing SNVs at those locations in figure 1.
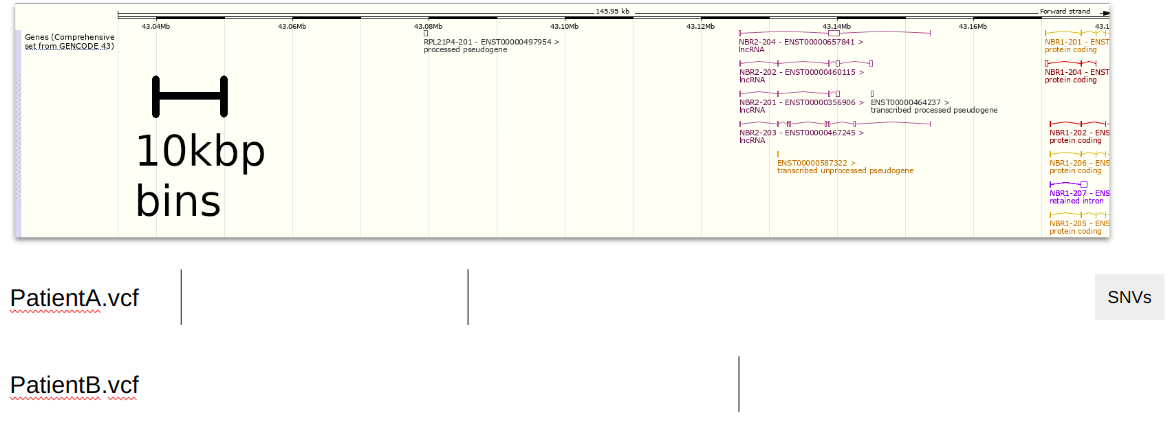
Since we use 10kbp bins, we create indexes for each SNV along those bins (see figure 2).
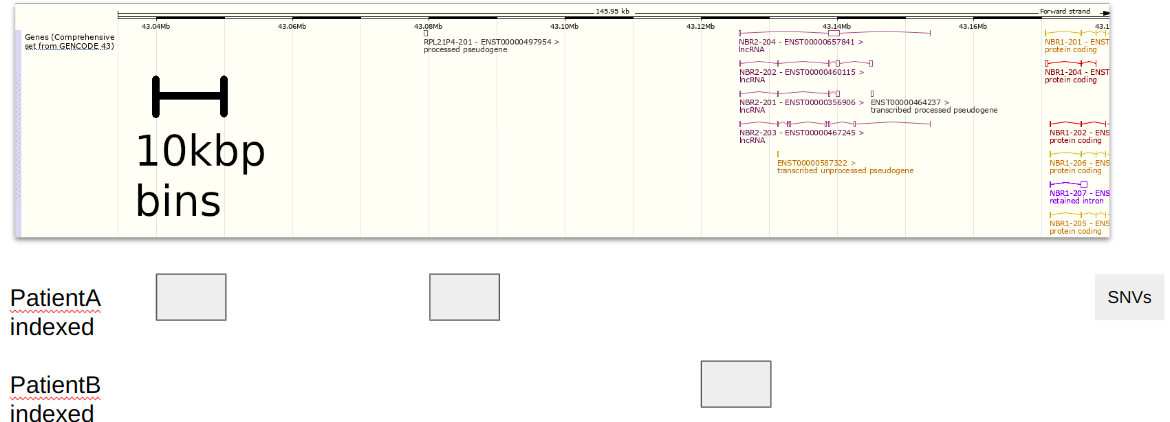
This index of bins->VCF, and the headers for each VCF, are now stored in the SQLite database for easier retrieval.
Data Search
When a search comes in, we:
- Transform the searched region into buckets, like above
- Check which files contain an entry in each of those buckets
- Run
pysamto grab only the parts of the VCF files that are relevant - Postprocess those files for the Beacon protocol using the headers for each file, also stored in SQLite
Suppose we have a request for a region covered by one of the VCFs above. First, this request is transformed into bins, and we search the index for each file covered by those bins (figure 3).

From the indexes, we know that PatientA.vcf has variants in this region. We can then grab the relevant segment via pysam, and combine it with header information for the metadata.
Frontend
There are two sections to the frontend: a summary landing page (figure 4) and a search page (figure 5).

There are many features to this search page:
- A sidebar to filter results, similar to how other genomics sites allow you to filter data, passing these queries to the backend to obtain a paginated list on the frontend.
- Data privacy is also respected – if a researcher does not have the credentials necessary to view the patients, counts are returned instead.
- As we implement the GA4GH HTSGet protocol, we are interoperable with software that can interface with it (e.g. IGV viewers)
Conclusion
CanDIG allows researchers to find genomic variants of interest in a performant manner, using binning and indexes to speed up searches over the entire genome. These searches are supported by a frontend that shows the data privacy aspect of CanDIG and implements GA4GH HTSGet.
Do you have any questions? Feel free to contact us at info@distributedgenomics.ca or on Twitter at @distribgenomics.