
CanDIG, is building the next generation of it’s federated solution for the analysis of privacy-sensitive genomic data across Canada. It will enable clinical researchers from distributed sites to examine and analyze quality data, including data from cancer trials, with no need for a central infrastructure to maintain or secure. This sharing of research-quality data between numerous cancer trials and treatment centers will help generate essential information that will aid in the efficient and effective diagnosis, treatment and follow-up of health conditions.
An introduction to mCODE
According to findings by the American Society of Clinical Oncologists, only about 3 percent of adult cancer patients in the US participate in clinical trials, the other 97% of the data located in Electronic Health Records (EHRs) in various institutions, which most often have systems that are incompatible with one another.
One solution includes implementing standards or a common data model to collect and share health information. Standards are fundamental for the meaningful exchange of health information because they provide a shared framework to allow consistency in the meaning and format of data during its collection, usage and exchange. Seamless and comprehensive data sharing is especially crucial for healthcare organizations and initiatives that depend on data exchange for the success of their programs, such as the Marathon of Hope Cancer Centers Network, a project currently supported by CanDIG.
The Marathon of Hope Cancer Centers Network, (MOHCCN), the largest of its kind in Canada, is a consortium of cancer treatment and research centers across Canada with a vision to create a high quality and shareable dataset consisting of 15,000 cases in the next five years and 100,000 cases within the decade. For the success of this project, the EHRs and systems in these distributed centers must enable interoperability, possibly by integrating a common data standard. This requires data to be collected using a framework that supports agreed-upon elements or a minimum set of data elements that adequately support the analysis and reporting of oncology-related research.
One data model the CanDIGv2 platform utilizes is called minimal common oncology data elements, or mCODE, which plays an important role in the MoHCCN This framework contains core clinical and genomic data elements for cancer care and research, as indicated by the conceptual framework in figure 1.
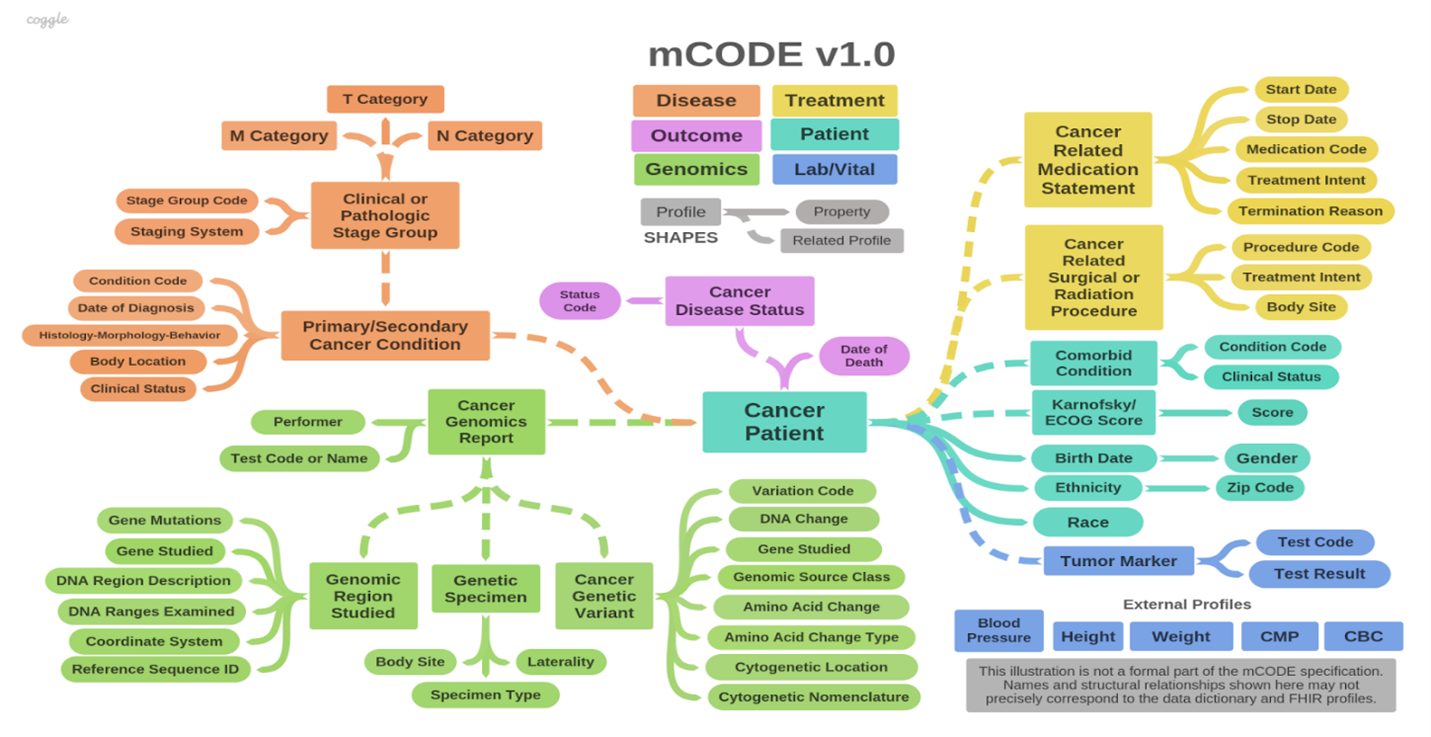
The colour schemata for this framework, illustrated in figure 1, represents the six major domains or groups that mCODE is organized: patient, disease, treatment, lab/vitals, genomics and outcome. The high-level domains are again categorized into 23 profiles composed of 93 data elements. In addition, the mCODE model contains five external profiles.
As an example of the decisions made when setting standards, while there are different methods of cancer staging, mCODE currently uses the TNM staging method (Figure 2). T stands for tumour and indicates the size of the tumor and whether it has grown to other parts of the body. N stands for node or lymph nodes and it describes whether the cancer has spread to the adjacent lymph nodes. Lastly, M, which stands for metastasis, indicates whether the cancer has spread to other parts of the body through the blood or lymphatic system.
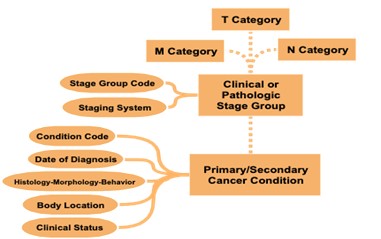
By utilizing the TNM system to stage cancer, clinicians and researchers can fast-track data sharing and aggregation to learn and gain new insights and improve patient outcomes. With the use of mCODE as a common data standard, the goal is to collect, use and share oncology data according to the elements specified by the model to enable interoperability between research and care institutions. To fully use the model, an mCODE data dictionary (Figure 3) has been developed, which describes each profile and its 93 different corresponding data elements. The data dictionary indicates which elements are required and which ones are required if known. Required if known is defined where the implementation shall provide “meaningful support” for the given elements.
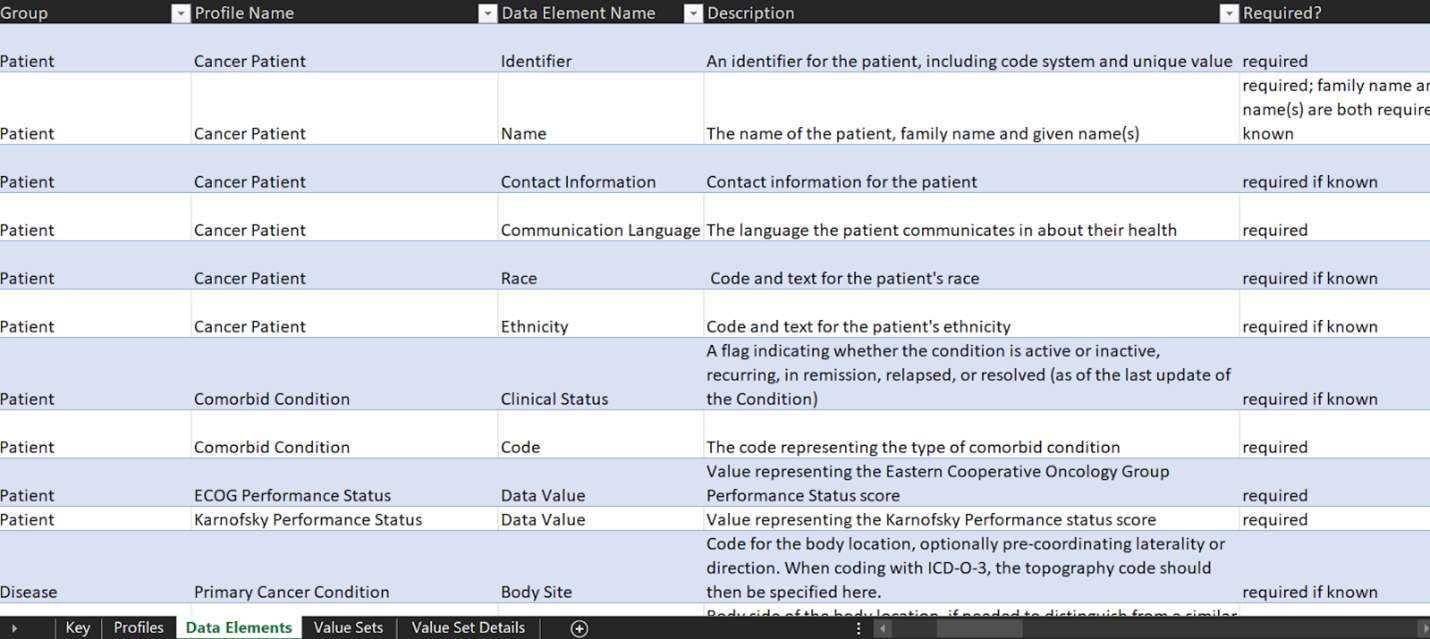
A required element, on the other hand, is where a data sender must provide each required element or an explicit “data absent reason” for each missing data item. A required element will have a cardinality of 1..1.
The data representation process
Figure 4 represents synthetic clinical data prepared for demonstration purposes from a hypothetical MoHCCN cohort called CORE. The sample clinical data illustrates data captured as “diagnosis” and contains specific values of data entered during the initial identification of cancer. Most of the data from research facilities come in the form of excel or google sheets, sometimes case report forms. The column headers are assigned a variable name which is different for each project. Figuring out the meaning of these variable names is a fundamental task required when doing mapping work. Variable names get stored in the dictionary of the data file, but there are occasions it might not be provided. In the event of any confusion surfacing with the meaning, a discussion with the data custodians is warranted.
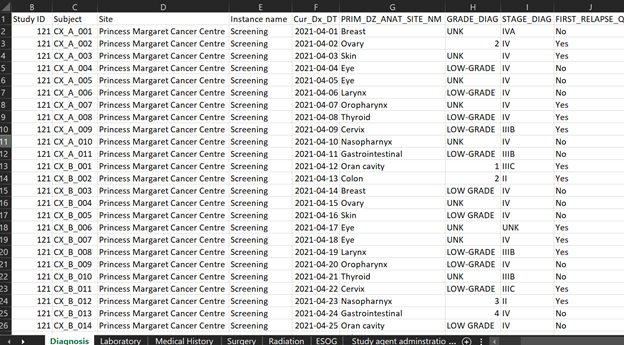
The first step of the mapping process is understanding the data: each variable name, what data it does and does not contain and the relationship across sections. The next step requires the manual transferring of data to mCODE according to the description in the data dictionary. At this stage, the aim is to map as much of the data to mCODE as possible. However, unless the data was originally collected using mCODE, it is common to have some level of data loss. This can be as a result of a number of factors, 1) the data to be mapped does not include the mCODE elements, 2) data available does not correspond to the mCODE description as per the data dictionary, or 3) the data shared contains additional information that is not captured by the data model. If we consider the previous excel dataset, we saw that the dataset contains cancer staging. Upon close examination at the completed mapping excel sheet (Figure 5), we can see rows 20 to 22 indicate N/A, meaning not available. This is because, as discussed previously, mCODE uses a TNM staging system, while the dataset contains another staging system not supported by mCODE. Cancer staging is a piece of important information in cancer research; however, this particular diagnosis dataset will not have staging data in mCODE. This could result in a significant data loss.
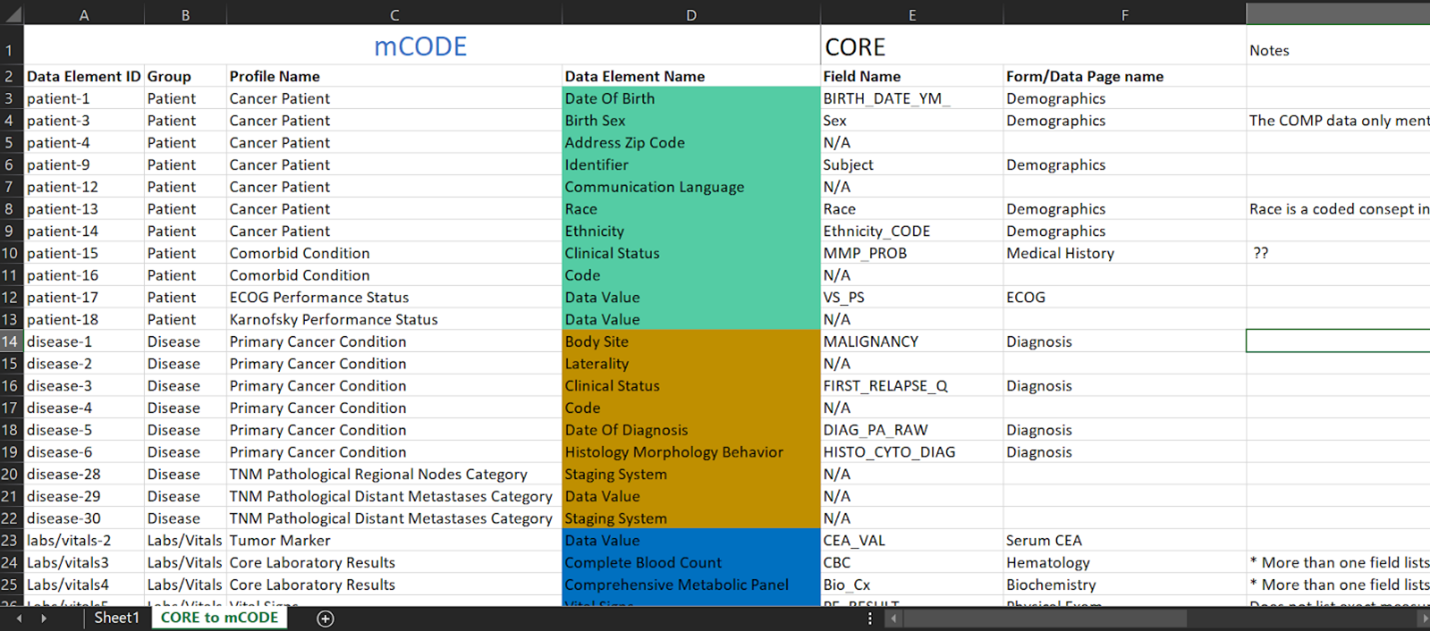
Separately, in the demographics section, row 4, where “Sex” from the CORE project dataset has been mapped to “birth sex” in mCODE. A remark in the notes section has been made to discuss with the data custodian to check if “sex” was recorded to mean birth sex. As we can see here with this example, in some instances, even those that may seem to be a simple modification may still require a detailed discussion with data custodians to assure accurate representation. After completing, the third step is to confirm with the data custodians that the mapping correctly represents the original data. At this stage, the data custodian is the primary person responsible to validate the meanings and terminologies used in the research project. Following the approval from the data custodian, the next step involves working with a developer to ingest the data to a server. Here, the person who completed the original mapping will provide final validation of the results and the representation is considered complete.
Extending mCODE, and combining mCODE data with data models used in other health domains, will make use of another data model which CanDIG will use, the Observational Medical Outcomes Partnership (OMOP).
OMOP will allow us to extend mCODE by supporting more comprehensive data. For example, data from a cancer project that we recently completed mapping to mCODE has additional data on laboratory tests such as urinalysis, thyroid function and coagulation tests and data on sites of metastasis that can not be represented in mCODE. We will continue using mCODE for MoHCCN projects and new patient cohorts that collect data using mCODE, and a corresponding mapping will be completed between the two data models, mCODE and OMOP, to facilitate extended representation of data elements.
Stay tuned for a blog post coming soon that dives deeper into OMOP!
Do you have any questions? Feel free to contact us at info@distributedgenomics.ca or on Twitter at @distribgenomics.